在阅读此文前,诚邀您请点点右上方的“关注”,既方便您进行讨论与分享,还能及时阅读最新内容,感谢您的支持。
新冠病毒暴发至今已有两年之久,但此刻仍不能放松警惕,新冠病毒已进化出多种“令人担忧的变异毒株”。
截至2021年8月,Delta变体都是有很高传播率的菌株;而Mu变体则是对血清中和的抗性最高。
世界卫生组织(WHO)在2021年11月26日举行的紧急会议中提到,最新出现的Omicron变异株正在全球肆虐,Omicron变体的突变比Delta多,甚至被认为是目前最危险的冠状病毒株,这引起了新一轮的感染高峰。
在病毒传播过程中,S蛋白中氨基酸的变化可能会显著改变病毒抗原性和中和抗体的效力。
随着SARS-CoV-2在世界各地的传播,S蛋白的突变不断被报道。
据GISAID数据库报告,SARS-CoV-2刺突蛋白中有930个自然发生的错义突变,SARS-CoV-2刺突蛋白中从ASP614到GLY614(D614G)的关键突变使SARS-CoV-2比原始菌株更具传染性。
然而,SARS-CoV-2的大多数疫苗、检测试剂和抗体都基于武汉参考菌株的S蛋白。
在其他冠状病毒中,错义突变被证明对MERS CoV和SARS CoV中的中和抗体具有耐药性。
就HIV而言,已知错义突变会影响包膜糖蛋白表达、病毒感染性、改变中和敏感性,并通过中和抗体产生耐药性。
SARS-CoV-2的S蛋白是受体利用的关键决定因素,S蛋白上的氨基酸突变甚至会增加病毒宿主的范围,在不同物种间进行传播。
SARS-CoV-2病毒通过S蛋白与人体受体ACE2结合,S蛋白由S1和S2亚基组成。
S1亚基有三个结构域:N端结构域(NTD),C端结构域(CTD)和受体结合结构域(RBD)。
而S蛋白的RBD区主要负责ACE2的识别,它是导致宿主感染的一个重要因素。
Omicron变异株最大的特点则是存在大量突变和免疫逃避,特别是S蛋白RBD区域的突变更加导致了逃避免疫的发生。
分子动力学是一种以分子力学为基础的计算机仿真模拟实验方法,主要用于模拟并观测分子在不同环境下的运动变化。
分子动力学模拟(MD)是分子模拟中最接近实验条件的模拟方法。
分子动力学模拟是目前为体系提供准确数据的最流行方法之一,随着新冠病毒不停突变,越来越多的人开始将分子动力学模拟方法运用到自己的蛋白体系研究当中,这为研究新冠蛋白结构的稳定性以及构象变化提供了帮助。
在分子动力学模拟之前一般需要做一些初始化操作,以计算某体系的结合自由能为例。
前期需要文件的处理,生成拓扑文件和坐标文件以及能量最小化。
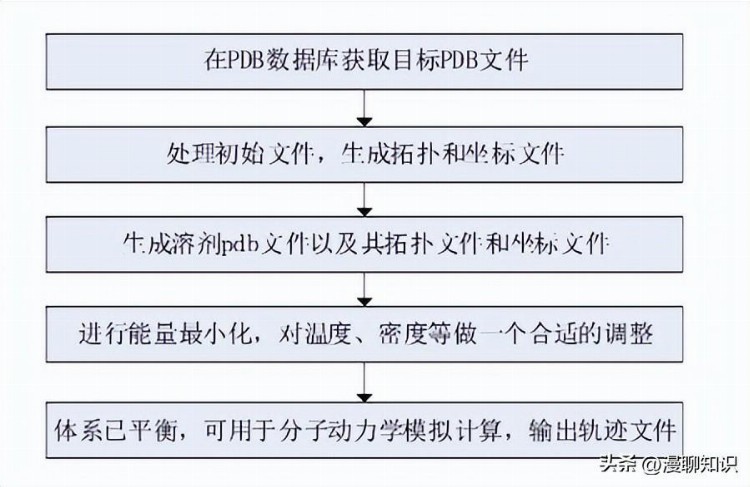
图1 计算结合自由能的一般流程
接下来,本文从分子动力学模拟在刺突蛋白和ACE2的结合以及刺突蛋白和中和抗体的结合中的应用等多个方面进行阐述,然后讨论目前刺突蛋白突变面临的挑战,分析并探讨刺突蛋白突变对突变体和人体细胞结合的发展情况。
用分子动力学模拟研究刺突蛋白和ACE2的结合
刺突蛋白和ACE2复合物的RMSD和RMSF
体系经过分子动力学模拟之后,一般会进行均方根偏差(RMSD)和均方根波动(RMSF)的计算,简单概括RMSD表示的是分子结构随着模拟时间变化的程度,而RMSF值表示的是分子中每个原子运动后与原始参考位置的波动程度。
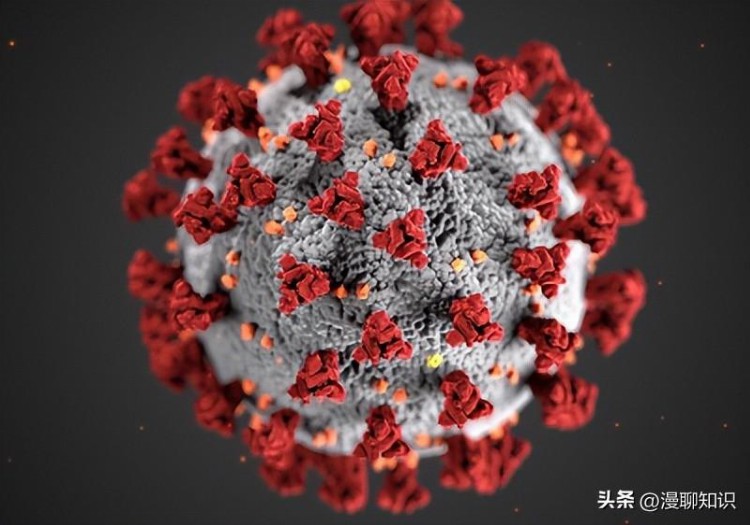
RMSD代表了模拟过程中蛋白系统的构象稳定性,因为它反映了蛋白质结构中原子相对于起始结构的平均位移,其计算公式如式(1)所示,N为原子总数,δi是指某一帧第i个原子相对于参考构像的位移偏移量。
而对其所有原子进行平方和除N平均再开方后,求得的为某一时刻这一帧的整体RMSD值。
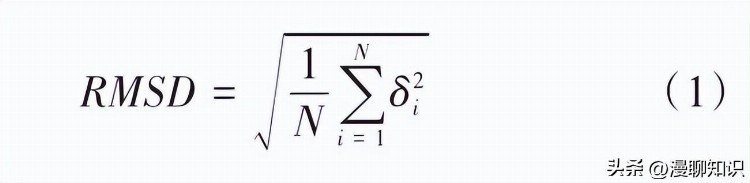
RMSF表示模拟后蛋白系统的残基位置与原始位置相比的波动程度,其计算公式如式(2)所示,其中引入了计算时间T,这里是得到时间段T所有时刻的位移平移量的平方和,最终求得的是某个原子在时间T内相对于初始结构的RMSF。
今年成都大学胡建平团队研究了SARS-CoV-2野生型和突变体Delta,Mu和Omicron与人体ACE2受体的结合情况,胡建平团队使用残基的基本结构取自PDB数据库,选取PDB ID分别为7KJ2和7V7S当作野生型和Delta变体的初始结构,然后在此基础上使用Pymol诱变工具和HadDock服务器进行突变,通过SWISS-MODEL服务器进行序列补全,就构建出了①四个全长S蛋白三聚体,包括WT、Delta、Mu和Omicron变体;②四种具有ACE2受体的三聚体复合物;③具有TMPRSS2的四个S蛋白三聚体对接复合体模型。
初始文件形成后开始进行分子动力学模拟,该团队使用ff14SB力场和Amber19软件包对12个S蛋白三聚体系统进行了MD模拟。
溶质被放置在边界为15.0Å的八面体盒中,其中溶剂效应由TIP3P水模型表征。
除了常规MD模拟的300 K温度设置外,还增加了310 K和320 K选项,以评估温度是否影响S蛋白构象。
在MD模拟之前,进行了最陡下降和溶质无约束最小化两步能量优化。
最后,在能量最小化后进行两阶段100 ns MD模拟。
第一阶段是5 ns溶质约束动力学,力常数为10 kcal·mol-1Å-2。
第二个是95 ns无约束生产模拟,其中采用SHAKE算法来防止涉及非氢原子的化学键的破坏。
通过其实验发现WT、Mu和Omicron变体在30 ns后趋于稳定,RMSD分别为4.61Å、4.84Å和5.15Å,而Delta在65 ns后才达到平衡,均方根偏差为6.75Å。
这表明这三种变体的构象稳定性不如WT,尤其是Delta。
为了分析不同温度对S蛋白结构稳定性的影响,在三种不同温度(即300 K、310 K和320 K)下进行了比较MD模拟,WT在300 K/310 K/320 K下的平均RMSD值依次为4.61Å/5.47Å/5.05Å,Delta、Mu和Omicron的RMSD值分别为6.75Å/5.34Å/5.24Å、4.84Å/4.59Å/5.09Å和5.15Å/4.8Å/4.26Å。
从结果可以看出Mu最为稳定而Omicron结构也比WT要稳定,但是Delta变种的稳定性要比WT差。
同样从RMSF的结果也可以看出几种变体稳定性从大到小依次是Mu、Omicron和Delta,但是都要比WT更加稳定。
刺突蛋白和ACE2复合物的结合自由能
结合自由能是查看体系结合情况的一个核心指标,平衡体系后使用脚本计算S蛋白-ACE2相互作用的能量项,该方法基于具有泊松-玻尔兹曼表面积的分子力学(MM-PBSA)方法。
ΔGSolv,Rec、ΔGSolv,Lig和ΔGSolv,Comp分别为气相中的受体、配体和复合物进入液相中时溶剂化能的变化;而ΔGSolv,bind和ΔGGas,bind分别为液相中和气相中的结合自由能。
理想情况下,如路径1使用在溶剂中的受体和配体结合直接计算结合自由能,但是在这些溶剂化状态的模拟中,总能量的波动将比结合能大一个数量级,因此结合能的计算需要非常长的时间才能收敛。
所以更有效的方法是把热力学循环下的各部分分开计算。
路径2的简化过程是先将受体和配体在气相中结合,然后再把结合物放在溶剂环境中。
从实验中可以看出,两者的路径始末状态相同,则图中的能量关系如下:
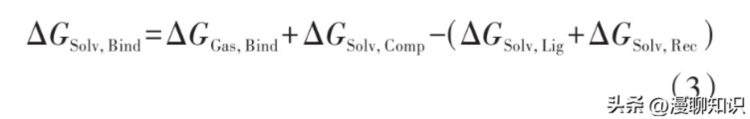
式(3)用能量类型进行表达可得:
式(4)中,焓变(ΔH)可以分解为气相能(ΔEMM)和溶剂化能(ΔGSol)[21],忽略熵(-TΔS)对ΔGBind的贡献,焓变公式表达为
式(5)中,气相能(ΔEMM)可被分解为静电项(ΔEele)、范德华项(ΔEvdW)和内能项(ΔEint)。
ΔEint内能由键能、键角能和二面角能组成。
ΔEMM分解表达如下式:
式(5)中的ΔGSol是极性溶剂化能(ΔGpb)和非极性溶剂化能(ΔGnp)之和:
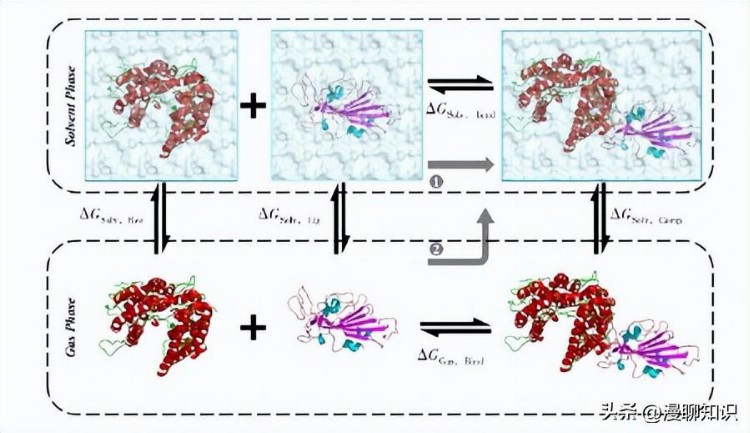
图2 MM⁃PBSA热力学循环情况
2021年,Zhou等计算了野生和突变体SARS-CoV-2 RBD-hACE2复合物的分子力结合自由能。
N439K-hACE2的结合能要高于野生型RBD-hACE2,野生型和突变型之间的静电能量有显著差异。
为了保证模拟的可靠性,Zhou等将MD模拟时间增加到200ns,200 nsMD模拟的结合能显示出与100 ns类似的结果,N439K-hACE2的结合能(-1597.25±179.57 KJ/mol)也高于野生型的结合能(-959.29±130.36 KJ/mol)。
同时,Zhou等用MMPBSA计算了RBM(thereceptor-bindingmotif)和hACE2之间的结合能,两个复合物的结合能分别为-860.94±99.45KJ/mol和-398.06±111.76KJ/mol。
比较RBD-hACE2和RBM-hACE2的结合自由能,发现能量的变化主要集中在RBM-hACE2区域,说明了S蛋白与hACE2的主要结合点位。
用分子动力学模拟研究刺突蛋白和中和抗体的结合
Kwarteng等使用GROMACS v2021和CH-ARMM36力场(模拟野生型和D614G S-蛋白系统)。
将蛋白质放置在一个立方体盒子中居中,置于盒子边缘1 nm处,使用TIP3P水模型溶剂化。
向系统中加入适当的离子以静电中和分子系统。
使用最速下降算法对系统进行能量最小化。
系统的蛋白质和非蛋白质组分独立耦合到vrescale恒温器和各向同性Berendsen算法进行压力耦合。
通过弱耦合至1 bar的参考压力维持压力,使用LINCS算法限制蛋白质内的键长。
NVT和NPT平衡总共进行了400 ps。
对所有分子系统进行了三次各自100 ns的独立模拟。
根据GROMACS指令求出野生型和D614G S-蛋白的平均RMSD值分别为1.09±0.04 nm和1.26±0.1 nm。
野生型和D614G S蛋白的结构相似性和构象稳定性存在显著差异,实验探究D614G S蛋白和野生型的结构相似性,并通过计算每个S蛋白结构域的RMSD来研究构象稳定性。
监测突变对S-蛋白构象稳定性的影响有助于研究病毒与中和抗体之间的相互作用。
计算了野生型、D614G和开放态S蛋白的NTD、RBD和S2结构域的RMSD[28]。
结果显示野生型和D614G S蛋白的RBD比开放态RBD具有更差的构象稳定性[29]。
与野生型和开放状态相比,D614G S蛋白的S2结构域显示出最高的RMSD值。
这表明,S蛋白骨架原子的平均位移主要由S2结构域的构象驱动。
突变对S蛋白结构域构象的影响在S2结构域中比NTD和RBD更显著。
总的来说,骨架RMSD和结构域特异性RMSD表明D614G S蛋白采用了独特的构象,但似乎比野生型构象更偏向于开放态构象,而比较野生型和D614G S蛋白的RBD区域会发现,突变会影响RBD的结构动力学。
分子动力学模拟在SARS-CoV-2中的其他应用
分子动力学模拟在病毒酶中的应用
由于该流行病仍在全球范围内蔓延,而负责病毒复制的酶又在病毒变异中起重要作用,因此采用模拟计算方法来确定这种酶的潜在抑制剂具有十分重要的意义。
Parise等研究了有吸引力的抗病毒核苷酸类似物RNA聚合酶(RdRp)链终结者抑制剂。
研究以分子动力学模拟为基础,探讨了内源性合成的三磷酸核苷(ddhCTP)与天然核碱基三磷酸环(CTP)在RdRp中结合的重要方面。
ddhCTP种类是毒蛇素抗病毒蛋白的产物,是先天免疫反应的一部分。
ddhCTP中核糖3"-OH的缺失可能对其抑制RdRp的机制有重要影响。
Parise等用实验方法建立了一个嵌入RdRp的RNA链的硅学模型,从低温电子显微镜结构开始,利用光谱法获得了RNA序列的信息。
该模型在稳定MD模拟计算后,获得的结果为三磷酸核苷的结合提供了更深入的见解,但是RdRp活性位点的分子机制仍然是难以捉摸的。
分子动力学模拟在发现药物靶点中的应用
最近,Yu等将TLR9人体蛋白列为研究治疗SARS-CoV-2药物靶点的目标蛋白,但此类治疗的特异性和疗效尚不清楚。
团队通过由相互作用化学物质搜索工具(STITCH)、京都基因和基因组百科全书(KEGG)、分子对接和病毒-宿主药物相互作用组映射组成的框架,对重组药物进行了研究。
以氯喹(CQ)和羟氯喹(HCQ)为探针,探索与SARS-CoV-2相关的相互作用网络。
47个药物靶点与SARS-CoV-2网络重叠,并富含TLR信号通路。
分子对接分析和分子动力学模拟确定了TLR9人体蛋白与CQ和HCQ的直接结合亲和力。
此外,该团队还建立了SARS-CoV-2-人类-药物-蛋白质相互作用图,不仅为SARS-CoV-2感染和病毒机制提供关键的见解,而且在发现新药物靶点方面起重要作用。
分子动力学模拟在疫苗设计中的应用
随着2019年冠状病毒大流行的暴发,Massomi等专注于寻找治愈冠状病毒疾病的解决方案上。
在所有的疫苗接种策略中,纳米颗粒疫苗已被证明可以使用于免疫系统,并在单次剂量中提供给免疫系统最佳的免疫状态。
铁蛋白是一种用于疫苗生产的纳米颗粒蛋白,目前已用于实验研究。
此外,糖基化在抗体和疫苗的设计中起着至关重要的作用,是开发有效疫苗的重要因素。
在这项计算研究中,铁蛋白纳米颗粒和糖基化是疫苗设计的两个独特方面,首次用于对改进的纳米颗粒疫苗进行建模。
在这方面,进行了分子建模和分子动力学模拟,以构SARS-CoV-2 RBD区-铁蛋白纳米颗粒疫苗的三个原子模型,包括未糖基化、糖基化和在铁蛋白-RBD界面用O-聚糖改进的疫苗。
结果表明,当聚糖添加到铁蛋白-RBD界面时,铁蛋白-RBSD复合物变得更稳定,并且可以实现该纳米颗粒的最佳性能。
如果通过实验验证,这些发现可以改进纳米颗粒的设计,以抵抗所有微生物感染。
RBD铁蛋白纳米颗粒是最有效的疫苗之一,可在单次剂量内提供对病毒的最佳免疫。
在这项工作中,分子建模和MD模拟将自组装铁蛋白纳米颗粒支架与糖基化相结合,以改进目前可用的铁蛋白RBD疫苗。
对SARS-CoV-2特异性铁蛋白–RBD纳米颗粒疫苗的三种状态进行了建模,包括未糖基化、糖基化和在铁蛋白–RBS界面用O-聚糖改进。
结果表明,糖基化通常保持纳米颗粒的稳定性。
通过引入包括额外糖基化位点的修饰环,稳定性显著提高。
这些发现可能对改进目前可用的铁蛋白–RBD纳米颗粒疫苗以及未来针对各种冠状病毒科的纳米颗粒疫苗设计至关重要。
面临的挑战
本文讲述了分子动力学模拟在SARS-CoV-2中的应用情况,特别是SARS-CoV-2 S蛋白突变前后和人体ACE2在SARS-CoV-2 S蛋白突变前后和中和抗体的结合情况,通过分子动力学模拟计算得出RMSD、RMSF和结合自由能这几项指标来说明体系的结合能力,从近些年的研究中可以发现,分子动力学模拟是分析SARS-CoV-2病毒情况很流行一种手段,但是SARS-CoV-2病毒的不断突变,数不胜数的突变体接踵而至,这为研究整个SARS-CoV-2病毒的突变情况带来了很大的挑战。
SARS-CoV-2的突变导致疫情不断严重,计算机模拟技术在S蛋白突变逃逸、基于酶结构的虚拟筛选、疫苗设计等方面均得到了广泛的应用。
其中MD模拟技术通过对野生型和突变型的蛋白的RMSD、RMSF、结合自由能分析,发现突变可导致S蛋白与ACE2蛋白结合能力增强,其与中和抗体的结合能力减弱,从原子水平上解释了免疫逃逸的机制。
计算机模拟技术与实验研究的进一步结合必将为新冠病毒的研究带来新的突破。
WANG J,XU X,ZHOU X,et al. Molecular simulation of SARS-CoV-2 spike protein binding to pangolin ACE2 or human ACE2 natural variants reveals altered susceptibility to infection[J]. The Journal of General Virology,2020,101(9):921-924.
HAN P,LI L,LIU S,et al. Receptor binding and complex structures of human ACE2 to spike RBD fr